Frontiers in Genetics
Introduction
efektiivinen populaatiokoko (ne) on tärkeä geneettinen parametri, joka arvioi geneettisen kulkeutumisen määrää populaatiossa. Ne-kokoon voivat vaikuttaa väestönlaskennan populaation koon (NC) vaihtelut, jalostussukupuolen suhde ja lisääntymismenestyksen vaihtelu.
Ne estimointi voidaan saavuttaa käyttämällä menetelmiä, jotka jakautuvat kolmeen menetelmäluokkaan: demografiseen, polveutuvaan tai merkkipohjaiseen (Flury et al., 2010). Polveutumistietoja on perinteisesti käytetty Ne-arvioiden saamiseksi kotieläimistä. Ne: n luotettavat arviot riippuvat kuitenkin siitä, onko sukutaulu täydellinen. Tällainen tietämystila on mahdollista joissakin kotimaisissa populaatioissa, joiden demografisia muuttujia on seurattu tarkasti riittävän monen sukupolven ajan. Käytännössä tätä lähestymistapaa sovelletaan kuitenkin vain muutamiin tapauksiin, joihin liittyy hyvin hoidettuja rotuja (Flury ym., 2010; Uimari ja Tapio, 2011).
yksi ratkaisu puutteellisen polveutumisen rajoittamisen poistamiseksi on arvioida Ne: n viimeaikainen kehitys genomitietojen avulla. Useat kirjoittajat ovat tunnustaneet, että Ne voitaisiin arvioida tiedoista yhteys epätasapaino (LD) (Sved, 1971; Hill, 1981). LD kuvaa eri lokusten alleelien ei-satunnaista assosiaatiota lokusten fysikaalisten paikkojen välisen rekombinaationopeuden funktiona genomissa. LD-allekirjoitukset voivat kuitenkin johtua myös demografisista prosesseista, kuten admixture ja genetic drift (Wright, 1943; Wang, 2005), tai prosesseista, kuten ”liftaamisesta” valikoivien pyyhkäisyjen aikana (Smith and Haigh, 1974) tai taustavalinnasta (Charlesworth et al., 1997). Tällaisissa skenaarioissa eri lokuksissa olevat alleelit liittyvät toisiinsa riippumatta niiden läheisyydestä perimässä. Olettaen, että populaatio on suljettu ja panmiktinen, neutraalien linkittämättömien lokusten välillä laskettu LD-arvo riippuu yksinomaan geneettisestä ajautumisesta (Sved, 1971; Hill, 1981). Tätä esiintymistä voidaan käyttää Ne: n ennustamiseen, koska LD: n varianssin (laskettu alleelitaajuuksilla) ja efektiivisen populaation koon välinen suhde tunnetaan (Hill, 1981).
Genotyypitystekniikan viimeaikaiset edistysaskeleet (esim., käyttämällä SNP helmi ryhmät kymmeniä tuhansia DNA-koettimia) ovat mahdollistaneet keräämisen valtavia määriä genomin laajuisia linkitys tiedot ihanteellinen arvioimiseksi ne karjan ja ihmisten muun muassa (esim., Tenesa et al., 2007; de Roos ym., 2008; Corbin et al., 2010; Uimari ja Tapio, 2011; Kijas ym., 2012). Kuitenkin, ohjelmisto työkalu, joka mahdollistaa estimointi Ne alkaen LD puuttuu, ja tutkijat tällä hetkellä luottaa yhdistelmä työkaluja manipuloida tietoja, päätellä LD, ja yleensä käyttää räätälöityjä skriptejä suorittaa asianmukaiset laskelmat ja arvioida Ne.
tässä kuvataan SNEP-ohjelmistotyökalua, jonka avulla voidaan arvioida Ne-trendejä sukupolvesta toiseen käyttäen SNP-tietoja, jotka korjaavat otoskokoa, vaiheistusta ja rekombinaationopeutta.
materiaalit ja menetelmät
SNEP: n käyttämä menetelmä LD: n laskemiseksi riippuu vaiheittaisten tietojen saatavuudesta. Kun vaihe on tiedossa, käyttäjä voi valita Hillin ja Robertsonin (1968) neliömäisen korrelaatiokertoimen, joka käyttää haplotyyppitaajuuksia määrittääkseen LD: n kunkin lokusparin välillä (yhtälö 1). Koska tunnettua vaihetta ei kuitenkaan ole, voidaan valita potenssiin Pearsonin lokusparien välinen tulomomenttikorrelaatiokerroin. Vaikka nämä kaksi lähestymistapaa eivät ole samat, ne ovat erittäin vertailukelpoisia (McEvoy et al., 2011):
missä pA ja pB ovat vastaavasti alleelien A ja B taajuudet kahdessa erillisessä lokuksessa (X, Y) n-yksilöistä mitattuna PAB on haplotyypin A-ja b-alleelien frekvenssi tutkitussa populaatiossa, x ja Y ovat genotyypin keskimääräiset frekvenssit ensimmäisessä ja toisessa lokuksessa, XI on yksilön i genotyyppi ensimmäisessä lokuksessa ja Yi on yksilön I genotyyppi toisessa lokuksessa. Yhtälö (2) korreloi genotyyppisen alleelin määrän haplotyypin taajuuksien sijaan, eikä siihen vaikuta kaksois heterotsygootit (tämä lähestymistapa johtaa samoihin estimaatteihin kuin –r2-vaihtoehto PLINKISSÄ).
SNEP arvioi historiallisen efektiivisen populaation koon, joka perustuu r2: n, Ne: n ja c: n (rekombinaationopeus) (yhtälö 3—Sved, 1971) suhteeseen ja jonka avulla käyttäjät voivat sisällyttää otoskokoon ja gameettisen vaiheen epävarmuuteen liittyviä korjauksia (yhtälö 4—Weir ja Hill, 1980):
missä n on tutkittavien yksilöiden lukumäärä, β = 2, kun gameettinen vaihe tunnetaan, ja β = 1, Jos sen sijaan vaihetta ei tunneta.
rekombinaationopeudesta voidaan päätellä useita approksimaatioita käyttäen vertailukohtana kahden lokuksen välistä fysikaalista etäisyyttä (δ) ja muuntamalla se linkkietäisyydeksi (d), joka kuvataan yleensä Mb(δ) ≈ cM(d). Pienille D: n arvoille jälkimmäinen approksimaatio on voimassa, mutta suuremmille D: n arvoille useiden rekombinaatiotapahtumien ja interferenssin todennäköisyys kasvaa, eikä myöskään karttaetäisyyden ja rekombinaationopeuden suhde ole lineaarinen, koska suurin mahdollinen rekombinaationopeus on 0, 5. Näin ollen, ellei käytetä hyvin lyhyttä δ, approksimaatio d ≈ c ei ole ihanteellinen (Corbin et al., 2012). Siksi toteutimme kartoitus toimintoja kääntää arvioitu d C, seuraavat Haldane (1919), Kosambi (1943), Sved (1971), ja Sved ja Feldman (1973). Aluksi SNEP päättelee kunkin SNPs-parin osalta D: n olevan suoraan verrannollinen δ: iin d = kδ: n mukaisesti, jossa k on käyttäjän määrittelemä rekombinaationopeuden arvo (oletusarvo on 10-8, kuten Mb = cM). Päätelty arvo δ voidaan sitten kohdistaa johonkin käytettävissä olevista kartoitustoiminnoista, jos käyttäjä sitä vaatii.
Ratkaisuyhtälö (3) Ne: lle ja sisältää kaikki kuvatut korjaukset, mahdollistaa Ne: n ennustamisen LD-datasta käyttäen (Corbin et al., 2012):
missä Nt on efektiivinen populaation koko t sukupolvia sitten laskettuna t = (2F(ct))-1 (Hayes et al., 2003), ct on rekombinaationopeus, joka on määritelty markkereiden väliselle fyysiselle etäisyydelle ja jota on valinnaisesti mukautettu edellä mainituilla kartoitusfunktioilla, r2adj on LD-arvo, joka on mukautettu otoskokoon ja α:= {1, 2, 2, 2} on mutaatioiden esiintymisen korjaus (Ohta ja Kimura, 1971). Siksi LD yli suurempia rekombinantti etäisyydet on informatiivinen viime Ne kun taas lyhyemmät etäisyydet antavat tietoa kaukaisempia aikoja menneisyydessä. Käytetään sidontajärjestelmää, jotta saadaan keskimääräiset R2-arvot, jotka kuvaavat LD: tä tietyillä lokusten välisillä etäisyyksillä. Käytössä oleva binning-järjestelmä käyttää seuraavaa kaavaa määritelläkseen kunkin bin minimi-ja maksimiarvot:
jossa bi (ℕ1) on kaikkien astioiden (totbiinien) i: s bin, minD ja maxD ovat vastaavasti minimi ja maksimi etäisyys snps: n ja X: n välillä on positiivinen reaaliluku (ℝ0), kun X on 1, etäisyyksien jakauma lokeroiden välillä on lineaarinen ja jokaisella lokerolla on sama etäisyysalue. Suuremmilla X-arvoilla etäisyyksien jakauma muuttuu, mikä mahdollistaa suuremman alueen viimeisissä astioissa ja pienemmän alueen ensimmäisissä astioissa. Jos tätä parametria muutetaan, käyttäjällä on riittävä määrä parivertailuja kunkin lokeron lopullisen Ne-estimaatin laatimiseksi.
esimerkkisovellus
testasimme SNEP: tä kahdella julkaistulla aineistolla, joita oli aiemmin käytetty kuvaamaan Ne: n trendejä ajan mittaan käyttäen LD: tä, Bos indicusta ja Ovis Ariesia . R2 arviot naudan aineistot saatiin kirjoittajat käyttäen GenABLE (Aulchenko et al., 2007) käyttäen alleelin minimitaajuutta (MAF) < 0, 01 ja säätämällä rekombinaationopeutta käyttäen Haldanen kartoitusfunktiota (Haldane, 1919). Lammasaineiston R2-estimaatit tekijät laskivat käyttäen PLINK-1,07: ää (Purcell et al., 2007), jonka Maf < 0,05 eikä muita korjauksia. Molemmissa autosomiaineistoissa R2-estimaatit, joissa otoskoko on korjattu yhtälöllä (4), jossa β = 2. Näissä vertailevissa analyyseissä SNEP: n komentorivi sisälsi samat parametrit, joita käytettiin julkaistuissa tiedoissa R2-estimaattien lisäksi, jotka laskettiin genotyyppiluvun ja SNEP: n uuden binning-strategian avulla.
tulokset
SNEP on monikielinen sovellus, joka on kehitetty C++: lla ja binääreillä yleisimmille käyttöjärjestelmille (Windows, OSX ja Linux) ladattavissa osoitteesta https://sourceforge.net/projects/snepnetrends/. Binääreihin liittyy käsikirja, jossa kuvataan SNEP: n vaiheittainen käyttö Ne: n suuntausten päättelemiseksi tässä kuvatulla tavalla. SNEP tuottaa tulostiedoston, jossa sarkain on rajattu sarakkeet, jotka osoittavat seuraavat tiedot jokaisesta Ne-estimointiin käytetystä lokerosta: niiden sukupolvien lukumäärä, joita lokero vastaa (esim., 50 sukupolvea sitten), vastaava Ne-estimaatti, kunkin SNPs-parin keskimääräinen etäisyys roskakorissa, keskimääräinen r2 ja R2: n keskihajonta roskakorissa sekä R2: n laskemiseen käytettyjen SNP: iden lukumäärä roskakorissa. Tämä tiedosto voidaan helposti tuoda Microsoft Excel, R tai muu ohjelmisto piirtää tuloksia. Tässä esitetyt tontit (Kuvat 1, 3) vastaavat tulostiedoston sukupolvien takaisia ja Ne-sarakkeita. Sarake, jossa on R2: n standardipoikkeama, annetaan käyttäjille, jotka voivat tarkastaa Ne-estimaatin varianssin kussakin lokerossa, erityisesti sellaisten lokeroiden osalta, jotka heijastavat vanhempia aika-arvioita ja jotka ovat vähemmän luotettavia, kun R2: n estimoinnissa Käytettyjen SNP: iden määrä pienenee.
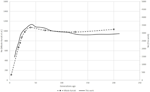
kuva 1. Vertailu Ne suuntauksia kuuden Sveitsin lammasrotuja mukaan Burren et al. (2014) (dashed lines) ja tämä teos (solid lines).
syöttötiedostojen edellyttämä muoto on tavallinen PLINK-muoto (ped-ja karttatiedostot) (Purcell et al., 2007). SNEP: n avulla käyttäjät voivat joko laskea LD: n tiedoista edellä kuvatulla tavalla tai käyttää mukautettua ennalta laskettua LD-matriisia estimoidakseen Ne yhtälön (5) avulla.
ohjelmointirajapinnan avulla käyttäjä voi hallita kaikkia analyysin parametreja, esim.snps: n välistä etäisyyttä bp: ssä ja analyysissä käytettävää kromosomijoukkoa (esim. 20-23). Lisäksi SNEP sisältää mahdollisuuden valita MAF-kynnysarvo (oletusarvo 0.05), koska on osoitettu, että MAF: n kirjanpidosta saadaan puolueettomia r2-estimaatteja otoskoosta riippumatta (Sved et al., 2008). SNEP: n monisäikeinen arkkitehtuuri mahdollistaa suurten datajoukkojen nopean laskemisen (testasimme jopa ~100K SNPs: ää yhdelle kromosomille), esimerkiksi tässä kuvatut BOS-tiedot analysoitiin yhdellä prosessorilla 2’43”: ssa, kahden suorittimen käyttö lyhensi aikaa 1’43”: een, neljä prosessoria vähensi analysointiaikaa 1’05”: een.
Zebu-esimerkki
zebu-analyysissä Snep: llä saatujen ne-käyrien muodot ja niiden julkaistut datasuuntaukset osoittivat saman liikeradan tasaisella laskulla noin 150 sukupolvea sitten, jota seurasi laajeneminen, jonka huippu oli noin 40 sukupolvea sitten ja joka päättyi jyrkkään laskuun viimeisimpiin sukupolviin verrattuna (Kuva 1). Vaikka molempien käyrien suuntaukset olivat samat, nämä kaksi lähestymistapaa johtivat erilaisiin Ne-estimaatteihin, ja SNEP: n arvot olivat noin kolme kertaa suurempia kuin alkuperäisessä paperissa. Vaikka yritimme käyttää tekijöiden parametreja analyyseissämme, jotkin erot olivat väistämättömiä, eli nautatiedon alkuperäisessä julkaisussa arvioitiin r2 erilaisella lähestymistavalla kuin SNEP: ssä. SNEP: llä tehdyt analyysit perustuivat genotyyppeihin, kun taas alkuperäinen analyysi perustui pääteltyihin kahteen lokuksen haplotyyppiin, jolloin julkaistut tiedot osoittivat odotetun r2: n olevan 0,32 minimietäisyydellä, kun meidän arviomme oli 0,23. Samoin Mbole-Kariuki et al. (2014) saatu Tausta tasolla r2 = 0.013 noin 2 Mb, kun meidän arvio samalla etäisyydellä oli 0.0035 (tietoja ei näy). Näin ollen, koska arviomme LD olivat jatkuvasti pienempiä kuin Mbole-Kariuki et al. (2014) on odotettavissa, että meidän Ne arviot olisi suurempi. Vaikka tämä havainto korostaa parametrien ja niiden raja-arvojen huolellisen valinnan tärkeyttä, on tärkeää korostaa, että vaikka Ne-arvojen absoluuttinen suuruus on erilainen, trendit ovat lähes identtiset.
esimerkki sveitsiläisistä lampaista
kuusi sveitsiläistä lammasrotua, jotka analysoitiin SNeP: llä, tuottivat vertailukelpoisia tuloksia kuin alkuperäisessä paperissa (kuva 2), enimmäkseen päällekkäisiä Ne-trendikäyriä (kuva 3). Yleinen suuntaus Ne: ssä oli kuitenkin laskussa nykyhetkeä kohti. SNEP tuotti hieman suuremmat Ne-arvot kaukaisemmalle menneisyydelle (700-800 sukupolvea). Tämä johtuu SNEP: ssä käytetystä erilaisesta binning-järjestelmästä, jonka avulla käyttäjä voi saada parivertailujen tasaisemman jakauman kunkin Binin sisällä (ts., SNP: n parivertailujen lukumäärä kussakin lokerossa on vertailukelpoinen). Ajanjaksolle, joka ulottuu yli 400 sukupolvea sitten, Burren et al. (2014) käytetään vain kolme Roskakorit niiden analyysi (keskitetty 400, 667, ja 2000 sukupolvet sitten), kun taas samaan aikaan span SNEP käytetään 5 Roskakorit useita parivertailuja riippuu alueen määritelty kaavoilla 6a, b. näin ollen, Burren ja kollegoiden lähestymistapa päättyy suurempi tiheys tietojen kuvataan uusimmat sukupolvet kuin kuvataan vanhimmat sukupolvet. Näin ollen pienempien säiliöiden käyttö lisää yleensä pienempien Ne-arvojen esiintymistä kussakin roskakorissa, mikä alentaa kunkin roskakorin Keskimääräistä Ne-arvoa. Lähimenneisyyden Ne-arvot, joita verrattiin 29. sukupolveen aiemmin, antoivat hyvin samanlaisia tuloksia. Suurin ero (50) saatiin SBS-rotuun.

kuva 2. Vertailu viime ne arvot laskettu 29. sukupolvi tässä työssä ja Burren et al. (2014) kuuden Sveitsin lammasrotuja.
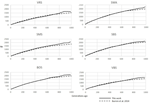
kuva 3. 250 viime sukupolven Ne-suuntausten vertailu Mbole-Kariuki et al: n saamissa SHZ-tiedoissa. (2014) (dashed line) ja käyttäen SNEP (solid line).
Keskustelu
niiden analysointi LD-tietojen avulla osoitettiin ensimmäisen kerran 40 vuotta sitten, ja sitä on sovellettu, kehitetty ja parannettu siitä lähtien (Sved, 1971; Hayes et al., 2003; Tenesa ym., 2007; de Roos ym., 2008; Corbin et al., 2012; Sved et al., 2013). Analysoitujen SNP-piirien perinteisesti pieni määrä ei ole enää rajoitus, sillä SNP-siruihin kuuluu erittäin suuri määrä SNP-siruja, jotka ovat saatavilla lyhyessä ajassa ja kohtuulliseen hintaan. Tämä on lisännyt menetelmän käyttöä, jota on sovellettu ihmisiin (Tenesa et al., 2007; McEvoy et al., 2011)sekä useille kesytetyille lajeille (England et al., 2006; Uimari ja Tapio, 2011; Corbin ym., 2012; Kijas et al., 2012). Näiden parannusten ohella metodologiset rajoitukset ovat tulleet ilmeisiksi, ja niitä on käsitelty tässä, ja suurin osa ponnisteluista on osoittanut, että viime aikojen tulokset on arvioitu oikein. Arvion kvantitatiivinen arvo riippuu kuitenkin suuresti otoksen koosta, LD-estimoinnin tyypistä ja binning-prosessista (Waples and Do, 2008; Corbin et al., 2012), kun taas sen laadullinen malli riippuu enemmän geneettisestä informaatiosta kuin tietojen manipuloinnista.
tähän mennessä tätä menetelmää on sovellettu käyttäen erilaisia ohjelmistoja, ei ole olemassa standardoitua lähestymistapaa tulosten bin ja jokainen tutkimus on sovellettu enemmän tai vähemmän mielivaltainen lähestymistapa, esim., binning sukupolven luokat aiemmin (Corbin et al., 2012), binning etäisyysluokille, joissa on vakioalue kullekin bin: lle (Kijas et al., 2012)tai binning per etäisyysluokat lineaarisesti, mutta suuremmilla astioilla uudemmille aikapisteille (Burren et al., 2014). Tietääksemme ainoa käytettävissä oleva ohjelmisto, joka arvioi Ne LD: n kautta, on NeEstimator (Do et al., 2014), päivitetty versio entisestä LDNE: stä (Waples and Do, 2008), joka mahdollistaa suurten aineistojen analysoinnin (50K SNPChip). Tärkeää on, että vaikka Snep keskittyy historiallisten uusien suuntausten arviointiin, Neestimatorin tavoitteena on tuottaa nykyaikaisia puolueettomia Ne-arvioita, jälkimmäistä olisi siksi pidettävä täydentävänä välineenä, kun väestökehitystä tutkitaan LD: n avulla.
käytimme SNEP: tä analysoimaan kahta aineistoa, joissa menetelmää on aiemmin sovellettu. Tulokset saimme lammas tiedot olivat sekä määrällisesti että laadullisesti vertailukelpoisia kuin saatu Burren et al. (2014), kun taas Zebu tiedot saimme Ne suuntaus arvio, joka vastasi läheisesti että Mbole-Kariuki et al. (2014) vaikka pistearviomme Ne olivat suurempia kuin kuvatut tiedot (Mbole-Kariuki et al., 2014). Ero näiden kahden tuloksen välillä johtuu siitä, että Burren ja kollegat tuottivat r2-estimaattinsa käyttäen PLINKIÄ (standardiohjelmistoa suuren mittakaavan SNP-tietojen manipulointia varten), joka käyttää samaa lähestymistapaa kuin Snep, kun taas Mbole-Kariuki ym. seurasi Hao et al. (2007) r2: n estimoinnin osalta. Eri estimaattien käyttö LD: lle on kriittistä Ne-käyrän kvantitatiiviselle aspektille, jossa Ne: n ja r2: n välisen hyperbolisen korrelaation vuoksi R2: n lasku sen alueella lähempänä arvoa 0 voi johtaa hyvin suureen muutokseen Ne-estimaateissa, kun taas estimaattien erot ovat vähemmän merkittäviä, kun r2-arvo on suuri eli lähempänä arvoa 1. Näin ollen, vaikka yhdessä aineistossa Ne-arvot poikkeavat huomattavasti toisistaan, molemmissa tapauksissa Ne-käyrät olivat päällekkäisiä alun perin julkaistujen kanssa.
kuten muut kirjoittajat jo ehdottivat, tällä menetelmällä saatujen määrällisten arvioiden luotettavuuteen on suhtauduttava varauksella, erityisesti uusimpiin ja vanhimpiin sukupolviin liittyvien Ne-arvojen osalta (Corbin et al., 2012)koska viime sukupolvien, suuria arvoja c ovat mukana, ei sovi teoreettisia vaikutuksia, että Hayes ehdotti arvioida muuttuja ne ajan (Hayes et al., 2003). Vanhimpien sukupolvien arviot saattavat myös olla epäluotettavia, koska koalitioteoria osoittaa, ettei SNP: tä voida luotettavasti ottaa 4NE-sukupolvien jälkeen menneisyydessä (Corbin et al., 2012). Lisäksi Ne-estimaatteihin ja erityisesti aikaisempiin sukupolviin liittyviin estimaatteihin vaikuttavat voimakkaasti tietojen manipulointitekijät, kuten MAF-ja alfa-arvojen valinta. Lisäksi käytetty sidontastrategia voi häiritä menetelmän yleistä tarkkuutta esimerkiksi silloin, kun kuhunkin lokeroon käytetään riittämätöntä määrää parivertailuja.
yksi menetelmän sovelluksista on rotujen demografioiden vertailu. Tässä tapauksessa Ne-käyrien muoto olisi optimaalinen väline eri väestöhistorioiden erottamiseen, enemmän kuin niiden numeeriset arvot, käyttämällä niitä kyseisen rodun tai lajin mahdollisena demografisena sormenjälkenä, ottaen kuitenkin huomioon, että mutaatio, migraatio ja valinta voivat vaikuttaa Ne-estimointiin LD: n kautta (Waples and Do, 2010). Lisäksi SNEP: n (ja muiden Ne: n arvioimiseen tarkoitettujen ohjelmistojen) kanssa analysoitujen tietojen huolellinen tarkastelu on erittäin tärkeää, koska sekoittavien tekijöiden, kuten admixture, esiintyminen voi johtaa ne: n puolueellisiin arvioihin (Orozco-terWengel and Bruford, 2014).
SNEP: n tavoitteena on näin ollen tarjota nopea ja luotettava työkalu, jolla voidaan soveltaa LD-menetelmiä Ne: n arvioimiseksi käyttämällä suuritehoisia genotyyppitietoja johdonmukaisemmin. Se mahdollistaa kaksi erilaista r2-estimointimenetelmää sekä mahdollisuuden käyttää ulkoisen ohjelmiston R2-estimaatteja. SNEP: n käyttö ei ylitä menetelmän ja sen taustalla olevan teorian rajoja, mutta se antaa käyttäjälle mahdollisuuden soveltaa teoriaa käyttäen kaikkia tähän mennessä ehdotettuja korjauksia.
Tekijäosuudet
MB suunnitteli ja kirjoitti ohjelmiston ja käsikirjoituksen. MB, MT ja POtW testasivat ohjelmiston ja tekivät analyysit. MT, POtW ja MWB tarkistivat käsikirjoituksen. Kaikki kirjoittajat hyväksyivät lopullisen käsikirjoituksen.
Eturistiriitalausunto
kirjoittajat toteavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, joita voitaisiin pitää mahdollisena eturistiriitana.
kiitokset
Kiitämme Christine Flurya lammastiedon antamisesta ja hyödyllisestä keskustelusta. Kiitämme myös kahta arvioijaa hyödyllisistä ehdotuksista tämän asiakirjan parantamiseksi. MB: tä tuki ohjelma Master and Back (Regione Sardegna).
Charlesworth, B., Nordborg, M., and Charlesworth, D. (1997). Paikallisen valinnan, tasapainoisen polymorfismin ja taustavalinnan vaikutukset geneettisen monimuotoisuuden tasapainomalleihin jakautuneissa populaatioissa. Genet. Res. 70, 155-174. doi: 10.1017/S0016672397002954
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Crow, J. F., and Kimura, M. (1970). Johdatus populaatiogenetiikan teoriaan. New York, NY: Harper ja Row.
Google Scholar
Ohta, T., and Kimura, M. (1971). Yhdistä epätasapaino kahden eriytyvän nukleotidikohdan välille mutaatioiden tasaisessa vuossa äärellisessä populaatiossa. Genetics 68, 571-580.
PubMed Abstract | Full Text/Google Scholar
Wright, S. (1943). Eristäminen etäisyyden mukaan. Genetics 28, 114-138.
PubMed Abstract / Full Text / Google Scholar