frontiere în genetică
Introducere
dimensiunea efectivă a populației (Ne) este un parametru genetic important care estimează cantitatea de drift genetic într–o populație și a fost descris ca dimensiunea unei populații idealizate Wright-Fisher care se așteaptă să producă aceeași valoare a unui parametru genetic dat ca și în populația studiată (Crow și Kimura, 1970). Dimensiunile Ne pot fi influențate de fluctuațiile dimensiunii populației recensământului (Nc), de raportul de sex de reproducere și de variația succesului reproductiv.
estimarea Ne poate fi realizată folosind abordări care se încadrează în trei categorii metodologice: demografice, bazate pe pedigree sau bazate pe markeri (Flury și colab., 2010). Datele Pedigree au fost utilizate în mod tradițional pentru a obține estimări Ne la animale. Cu toate acestea, estimările fiabile ale Ne depind de faptul că pedigree-ul este complet. Această stare de cunoaștere este fezabilă în unele populații interne, ale căror parametri demografici au fost monitorizați cu exactitate pentru un număr suficient de mare de generații. Cu toate acestea, în practică, aplicabilitatea acestei abordări rămâne limitată la câteva cazuri care implică rase bine gestionate (Flury și colab., 2010; Uimari și Tapio, 2011).
o soluție pentru a depăși limitarea unui pedigree incomplet este estimarea tendinței recente În Ne folosind date genomice. Mai mulți autori au recunoscut că Ne ar putea fi estimat din informații despre dezechilibrul de legătură (LD) (Sved, 1971; Hill, 1981). LD descrie asocierea non-aleatorie a alelelor în diferite loci în funcție de rata de recombinare între pozițiile fizice ale locilor din genom. Cu toate acestea, semnăturile LD pot rezulta și din procese demografice, cum ar fi amestecul și deriva genetică (Wright, 1943; Wang, 2005), sau prin procese precum „autostopul” în timpul măturărilor selective (Smith și Haigh, 1974) sau selecția fundalului (Charlesworth și colab., 1997). În astfel de scenarii, alelele la diferite loci devin asociate independent de apropierea lor în genom. Presupunând că o populație este închisă și panmictică, valoarea LD calculată între loci neutri nelegați depinde exclusiv de deriva genetică (Sved, 1971; Hill, 1981). Această apariție poate fi utilizată pentru a prezice Ne datorită relației cunoscute dintre varianța în LD (calculată folosind frecvențele alelelor) și dimensiunea efectivă a populației (Hill, 1981).
progrese recente în tehnologia de genotipare (de ex., folosind matrice de bile SNP cu zeci de mii de sonde ADN) au permis colectarea unor cantități mari de date de legătură la nivel de genom, ideale pentru estimarea Ne la animale și oameni, printre altele (de exemplu, Tenesa și colab., 2007; De Roos și colab., 2008; Corbin și colab., 2010; Uimari și Tapio, 2011; Kijas și colab., 2012). Cu toate acestea, un instrument software care permite estimarea Ne din LD lipsește, iar cercetătorii se bazează în prezent pe o combinație de instrumente pentru a manipula datele, a deduce LD și tind să utilizeze scripturi personalizate pentru a efectua calculele și estimarea ne corespunzătoare.
aici descriem SNeP, un instrument software care permite estimarea tendințelor Ne de-a lungul generației folosind date SNP care corectează dimensiunea eșantionului, fazarea și rata de recombinare.
materiale și metode
metoda SNeP folosește pentru a calcula LD depinde de disponibilitatea datelor pe etape. Când faza este cunoscută, utilizatorul poate selecta Hill și Robertson (1968) coeficient de corelație pătrat care folosește frecvențele haplotipului pentru a defini LD între fiecare pereche de loci (ecuația 1). Cu toate acestea, în absența unei faze cunoscute, coeficientul de corelație produs-moment pătrat al lui Pearson între perechi de loci poate fi selectat. Deși aceste două abordări nu sunt aceleași, ele sunt foarte comparabile (McEvoy și colab., 2011):
unde pA și pB sunt respectiv frecvențele alelelor A și B la două loci separate (X), Y) măsurată pentru n indivizi, PAB este frecvența haplotipului cu alele a și B în populația studiată, X și Y sunt frecvențele medii ale genotipului pentru primul și respectiv al doilea Locus, XI este genotipul individului i la primul Locus și Yi este genotipul individului i la al doilea locus. Ecuația (2) corelează numărul de alele genotipice în locul frecvențelor haplotipului și nu este influențată de heterozigoți dubli (această abordare are ca rezultat aceleași estimări ca opțiunea –r2 din PLINK).SNeP estimează dimensiunea istorică efectivă a populației pe baza relației dintre R2, Ne și C (rata de recombinare), (ecuația 3-Sved, 1971) și permite utilizatorilor să includă corecții pentru dimensiunea eșantionului și incertitudinea fazei gametice (ecuația 4-Weir și Hill, 1980):
unde N este numărul de persoane prelevate din eșantioane, 0% dacă faza gametică este cunoscută și 1% dacă faza gametică nu este cunoscută.
Mai multe aproximări sunt utilizate pentru a deduce rata de recombinare folosind distanța fizică (XV) între doi loci ca referință și traducând-o în Distanța de legătură (d), care este de obicei descrisă ca Mb(XV) cm(d). Pentru valorile mici ale lui d, această din urmă aproximare este valabilă, dar pentru valorile mai mari ale lui d probabilitatea unor evenimente multiple de recombinare și interferențe crește, în plus relația dintre distanța hărții și rata de recombinare nu este liniară, deoarece rata maximă de recombinare posibilă este de 0,5. Astfel, cu excepția cazului în care se utilizează foarte scurt, aproximarea D-C nu este ideală (Corbin și colab., 2012). Prin urmare, am implementat funcții de cartografiere pentru a traduce d estimat în c, după Haldane (1919), Kosambi (1943), Sved (1971) și Sved și Feldman (1973). Inițial SNeP deduce d pentru fiecare pereche de SNP-uri ca fiind direct proporțională cu XV în conformitate cu D = K, unde K este o valoare a ratei de recombinare definită de utilizator (valoarea implicită este de 10-8 ca în Mb = cM). Valoarea dedusă a lui hectolix poate fi apoi supusă uneia dintre funcțiile de cartografiere disponibile, dacă utilizatorul solicită acest lucru.
rezolvarea ecuației (3) Pentru Ne și includerea tuturor corecțiilor descrise, permite predicția Ne din datele LD folosind (Corbin și colab., 2012):
unde Nt este dimensiunea efectivă a populației t generații în urmă calculată ca t = (2f(ct))-1 (Hayes și colab., 2003), ct este rata de recombinare definită pentru o distanță fizică specifică între markeri și ajustată opțional cu funcțiile de cartografiere menționate mai sus, r2adj este valoarea LD ajustată pentru dimensiunea eșantionului și a lui ecuot:= {1, 2, 2.2} Este o corecție pentru apariția mutațiilor (Ohta și Kimura, 1971). Prin urmare, LD pe distanțe recombinante mai mari este informativ pe Ne recent, în timp ce distanțele mai scurte oferă informații despre perioade mai îndepărtate din trecut. Un sistem de binning este implementat pentru a obține valori medii r2 care reflectă LD pentru distanțe specifice inter-locus. Sistemul de binning implementat utilizează următoarea formulă pentru a defini valorile minime și maxime pentru fiecare bin:
în cazul în care bi (XV1) este al I-lea bin din numărul total de containere (totbins), minD, și maxD sunt, respectiv, minim și maxim distanța dintre SNP-uri și X este un număr real pozitiv (07) când X este egal cu 1, distribuția distanțelor dintre coșuri este liniară și fiecare coș are același interval de distanță. Pentru valori mai mari ale lui x, distribuția distanțelor se modifică, permițând o gamă mai mare pe ultimele coșuri și o gamă mai mică pe primele coșuri. Variația acestui parametru permite utilizatorului să aibă un număr suficient de comparații în perechi pentru a contribui la estimarea finală Ne pentru fiecare coș.
exemplu de aplicație
am testat SNeP cu două seturi de date publicate care au fost utilizate anterior pentru a descrie tendințele în Ne în timp folosind LD, Bos indicus și Ovis aries . Estimările r2 pentru seturile de date pentru bovine au fost obținute de autori folosind GenABLE (Aulchenko și colab., 2007) folosind o frecvență minimă a alelei (MAF) < 0,01 și ajustarea ratei de recombinare folosind funcția de cartografiere Haldane (Haldane, 1919). Estimările R2 ale datelor ovinelor au fost calculate de autori folosind PLINK-1.07 (Purcell și colab., 2007), cu un MAF < 0,05 și fără alte corecții. Pentru ambele seturi de date autozomale, estimările R2 au fost corectate pentru mărimea eșantionului utilizând ecuația (4) cu ecuația x = 2. Pentru aceste analize comparative, linia de comandă SNeP a inclus aceiași parametri utilizați pentru datele publicate, în afară de estimările r2, calculate prin numărul de genotipuri și utilizarea noii Strategii de binning a SNeP.
rezultate
SNeP este o aplicație multithreaded dezvoltată în C++ și binare pentru cele mai comune sisteme de operare (Windows, OSX și Linux) pot fi descărcate de lahttps://sourceforge.net/projects/snepnetrends/. Binarele sunt însoțite de un manual care descrie utilizarea pas cu pas a SNeP pentru a deduce tendințele în Ne așa cum este descris aici. SNeP produce un fișier de ieșire cu coloane delimitate de file care arată următoarele pentru fiecare coș care a fost utilizat pentru a estima Ne: Numărul de generații din trecut la care corespunde coșul (de ex., Acum 50 de generații), estimarea ne corespunzătoare, distanța medie dintre fiecare pereche de SNP-uri din Coș, media R2 și deviația standard a r2 din coș și numărul de SNP-uri utilizate pentru a calcula r2 în Coș. Acest fișier poate fi importat cu ușurință în Microsoft Excel, R sau alt software pentru a trasa rezultatele. Parcelele prezentate aici (Figurile 1, 3) corespund coloanelor generațiilor în urmă și Ne din fișierul de ieșire. Coloana cu deviația standard r2 este furnizată utilizatorilor pentru a inspecta varianța estimării Ne în fiecare coș, în special pentru acele coșuri care reflectă estimări de timp mai vechi și care sunt mai puțin fiabile pe măsură ce numărul de SNP-uri utilizate pentru estimarea r2 devine mai mic.
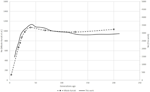
Figura 1. Compararea tendințelor Ne a șase rase de oi elvețiene conform Burren și colab. (2014) (linii punctate) și această lucrare (linii solide).
formatul necesar pentru fișierele de intrare este formatul PLINK standard (fișiere PED și map) (Purcell și colab., 2007). SNeP permite utilizatorilor fie să calculeze LD pe datele descrise mai sus, fie să utilizeze o matrice LD precalculată personalizată pentru a estima Ne folosind ecuația (5).
interfața software permite utilizatorului să controleze toți parametrii analizei, de exemplu, distanța dintre SNP în bp și setul de cromozomi utilizați în analiză (de exemplu, 20-23). În plus, SNeP include opțiunea de a alege un prag MAF (implicit 0.05), deoarece s-a demonstrat că contabilizarea MAF are ca rezultat estimări R2 imparțiale, indiferent de mărimea eșantionului (Sved și colab., 2008). Arhitectura multithreaded SNeP permite calcularea rapidă a seturilor de date mari (am testat până la ~100k SNP pentru un singur cromozom), de exemplu datele BOS descrise aici au fost analizate cu un procesor în 2’43”, utilizarea a două procesoare a redus timpul la 1’43”, patru procesoare au redus timpul de analiză la 1’05”.
exemplu Zebu
pentru analiza zebu, formele curbelor Ne obținute cu SNeP și tendințele datelor publicate au arătat aceeași traiectorie cu un declin lin până în urmă cu aproximativ 150 de generații, urmată de o expansiune cu un vârf în urmă cu aproximativ 40 de generații și care se încheie cu un declin abrupt pe cele mai recente generații (Figura 1). Cu toate acestea, în timp ce tendințele în ambele curbe au fost aceleași, cele două abordări au dus la estimări Ne diferite, valorile SNeP fiind de aproximativ trei ori mai mari decât cele din lucrarea originală. În timp ce am încercat să folosim parametrii autorilor în analizele noastre, unele diferențe au fost inevitabile, adică publicarea inițială a datelor despre bovine estimate r2 cu o abordare diferită față de cea implementată în SNeP. Analizele cu SNeP s-au bazat pe genotipuri, în timp ce analiza inițială s-a bazat pe două haplotipuri locus, ceea ce duce la datele publicate care arată un R2 așteptat de 0,32 la distanța minimă, în timp ce estimările noastre au fost de 0,23. În mod similar, Mbole-Kariuki și colab. (2014) a obținut un nivel de fundal r2 = 0,013 în jur de 2 Mb, în timp ce estimarea noastră la aceeași distanță a fost 0.0035 (datele nu sunt afișate). În consecință, deoarece estimările noastre despre LD au fost în mod constant mai mici decât Mbole-Kariuki și colab. (2014) este de așteptat ca estimările noastre Ne să fie mai mari. Deși această observație evidențiază importanța unei alegeri atente a parametrilor și a pragurilor acestora, este important să subliniem că, deși magnitudinea absolută a valorilor Ne este diferită, tendințele sunt aproape identice.
exemplu ovine elvețiene
cele șase rase ovine elvețiene analizate cu SNeP au produs rezultate comparabile cu cele din lucrarea originală (Figura 2), cu curbe de tendință ne în mare parte suprapuse (Figura 3). Cu toate acestea, tendința generală În Ne a arătat o scădere spre prezent. SNeP a produs valori ușor mai mari ale Ne pentru trecutul mai îndepărtat (700-800 generații). Acest lucru se datorează sistemului de binning diferit utilizat în SNeP, care permite utilizatorului să obțină o distribuție mai uniformă a comparațiilor în perechi în fiecare coș (adică., numărul de comparații SNP în perechi în fiecare coș este comparabil). Pentru intervalul de timp care se extinde dincolo de 400 de generații în urmă, Burren și colab. (2014) au folosit doar trei coșuri în analiza lor (centrată la 400, 667 și 2000 de generații în urmă), în timp ce pentru același interval de timp SNeP a folosit 5 coșuri cu un număr de comparații în perechi dependente de intervalul definit cu formulele 6a,b. în consecință, abordarea lui Burren și a colegilor se încheie cu o densitate mai mare de date care descriu cele mai recente generații decât descrierea celor mai vechi generații. Prin urmare, utilizarea mai puține coșuri tinde să crească prezența unor valori mai mici de Ne în fiecare coș, scăzând în consecință valoarea medie Ne pentru fiecare coș. Valorile Ne pentru trecutul recent, comparativ cu cea de-a 29-a generație din trecut, au dat rezultate foarte similare. Cea mai mare diferență (50) a fost obținută pentru rasa SBS.

Figura 2. Comparație între valorile Ne recente calculate la A 29-a generație în această lucrare și Burren și colab. (2014) pentru șase rase de oi elvețiene.
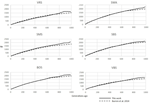
Figura 3. Compararea tendințelor Ne pentru ultimele 250 de generații în datele SHZ obținute de Mbole-Kariuki și colab. (2014) (linie punctată) și folosind SNeP (linie solidă).
discuție
analiza Ne folosind datele LD a fost demonstrată pentru prima dată acum 40 de ani și a fost aplicată, dezvoltată și îmbunătățită de atunci (Sved, 1971; Hayes și colab., 2003; Tenesa și colab., 2007; De Roos și colab., 2008; Corbin și colab., 2012; Sved și colab., 2013). Numărul tradițional mic de SNP-uri analizate nu mai este o limitare, deoarece cipurile SNP cuprind un număr extrem de mare de SNP-uri, disponibile într-un timp scurt și la un preț rezonabil. Acest lucru a stimulat utilizarea metodei, care a fost aplicată oamenilor (Tenesa și colab., 2007; McEvoy și colab., 2011), precum și la mai multe specii domesticite (Anglia și colab., 2006; Uimari și Tapio, 2011; Corbin și colab., 2012; Kijas și colab., 2012). Odată cu aceste îmbunătățiri, limitările metodologice au devenit evidente și au fost abordate aici, majoritatea eforturilor indicând estimarea corectă a Ne recente. Cu toate acestea, valoarea cantitativă a estimării depinde în mare măsură de dimensiunea eșantionului, de tipul estimării LD și de procesul de binning (Waples and Do, 2008; Corbin și colab., 2012), în timp ce modelul său calitativ depinde mai mult de informațiile genetice decât de manipularea datelor.
până în prezent, această metodă a fost aplicată folosind o varietate de software, nu există o abordare standardizată pentru a elimina rezultatele și fiecare studiu a aplicat o abordare mai mult sau mai puțin arbitrară, de exemplu, binning pentru clasele de generație în trecut (Corbin și colab., 2012), binning pentru clasele la distanță cu un interval constant pentru fiecare coș (Kijas și colab., 2012) sau binning pe clase de distanță într-un mod liniar, dar cu coșuri mai mari pentru punctele de timp mai recente (Burren și colab., 2014). Din cunoștințele noastre, singurul software disponibil care estimează Ne prin LD este NeEstimator (Do și colab., 2014), o versiune actualizată a fostului Ldne (Waples and Do, 2008) care permite analiza unui set de date mare (as 50k SNPChip). Important, în timp ce SNeP se concentrează pe estimarea tendințelor Ne istorice, scopul NeEstimator este de a produce estimări ne imparțiale contemporane, acestea din urmă ar trebui, prin urmare, considerate un instrument complementar în timp ce investighează demografia prin LD.
am folosit SNeP pentru a analiza două seturi de date în care metoda a fost aplicată anterior. Rezultatele obținute la ovine au fost comparabile cantitativ și calitativ cu cele obținute de Burren și colab. (2014), în timp ce pentru datele Zebu am obținut o estimare a tendințelor Ne care se potrivea îndeaproape cu cea a lui Mbole-Kariuki și colab. (2014) deși estimările noastre punctuale ale Ne au fost mai mari decât cele descrise pentru date (Mbole-Kariuki și colab., 2014). Discrepanța dintre aceste două rezultate reflectă faptul că Burren și colegii lor și-au produs estimările r2 folosind PLINK (software-ul standard pentru manipularea datelor SNP la scară largă) care utilizează aceeași abordare utilizată pentru a estima R2 de SNeP, în timp ce Mbole-Kariuki și colab. urmat Hao și colab. (2007) pentru estimarea r2. Utilizarea diferitelor estimări pentru LD este critică pentru aspectul cantitativ al curbei Ne, unde datorită corelației hiperbolice dintre Ne și r2, o scădere a r2 pe intervalul său mai aproape de 0 poate duce la o schimbare foarte mare a estimărilor Ne, în timp ce diferențele de estimări sunt mai puțin semnificative atunci când valoarea r2 este mare, adică mai aproape de 1. Prin urmare, deși într-unul dintre seturile de date valorile Ne au fost substanțial diferite, în ambele cazuri curbele Ne s-au suprapus cu cele publicate inițial.
după cum au sugerat deja alți autori, fiabilitatea estimărilor cantitative obținute cu această metodă trebuie luată cu precauție, în special pentru valorile ne legate de cele mai recente și cele mai vechi generații (Corbin și colab., 2012) deoarece pentru generațiile recente sunt implicate valori mari ale c, care nu se potrivesc cu implicațiile teoretice pe care Hayes le-a propus să estimeze o variabilă Ne în timp (Hayes și colab., 2003). Estimările pentru cele mai vechi generații ar putea fi, de asemenea, nesigure, deoarece teoria coalescentă arată că niciun SNP nu poate fi eșantionat în mod fiabil după generațiile 4Ne din trecut (Corbin și colab., 2012). În plus, estimările Ne, și în special cele legate de generațiile viitoare în trecut, sunt puternic afectate de factorii de manipulare a datelor, cum ar fi alegerea valorilor MAF și alfa. În plus, strategia de binning aplicată poate interfera cu precizia generală a metodei, de exemplu în cazul în care un număr insuficient de comparații în perechi sunt utilizate pentru a popula fiecare coș.
una dintre aplicațiile metodei este de a compara demografiile rasei. În acest caz, forma curbelor Ne ar fi instrumentul optim pentru diferențierea diferitelor istorii demografice, mai mult decât valorile lor numerice, prin utilizarea lor ca amprentă demografică potențială pentru acea rasă sau specie, luând în considerare totuși că mutația, migrația și selecția pot influența estimarea Ne prin LD (Waples și Do, 2010). În plus, o analiză atentă a datelor analizate cu SNeP (și alte programe software pentru estimarea Ne) este foarte importantă, deoarece prezența unor factori confuzi, cum ar fi amestecul, poate duce la estimări părtinitoare ale Ne (Orozco-terWengel și Bruford, 2014).
scopul SNeP este, prin urmare, de a oferi un instrument rapid și fiabil pentru a aplica metode LD pentru a estima Ne folosind date genotipice de mare capacitate într-un mod mai consistent. Permite două abordări diferite de estimare r2 plus opțiunea de a utiliza estimări r2 din software extern. Utilizarea SNeP nu depășește limitele metodei și teoria din spatele ei, dar permite utilizatorului să aplice teoria folosind toate corecțiile sugerate până în prezent.
contribuții autor
MB conceput și a scris software-ul și manuscrisul. MB, MT și POtW au testat software-ul și au efectuat analizele. MT, POtW și MWB au revizuit manuscrisul. Toți autorii au aprobat manuscrisul final.
Declarație privind conflictul de interese
autorii declară că cercetarea a fost realizată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
mulțumiri
îi mulțumim lui Christine Flury pentru furnizarea datelor despre oi și pentru discuții utile. De asemenea, mulțumim celor doi recenzori pentru sugestiile utile de îmbunătățire a acestei lucrări. MB a fost susținut de programul Master and Back (Regione Sardegna).Charlesworth, B., Nordborg, M. și Charlesworth, D. (1997). Efectele selecției locale, polimorfismului echilibrat și selecției de fond asupra modelelor de echilibru ale diversității genetice în populațiile subdivizate. Genet. Res. 70, 155-174. doi: 10.1017 / S0016672397002954
PubMed Abstract | Full Text | CrossRef Full Text/Google Scholar
Crow, J. F. și Kimura, M. (1970). O introducere în teoria geneticii populației. New York, NY: Harper și Row.
Google Scholar
Ohta, T. și Kimura, M. (1971). Dezechilibru de legătură între două situsuri nucleotidice segregante sub fluxul constant de mutații într-o populație finită. Genetica 68, 571-580.
PubMed Abstract | Text Complet/Google Scholar
Wright, S. (1943). Izolarea prin distanță. Genetica 28, 114-138.
PubMed rezumat / text integral / Google Scholar