Frontiers in Genetics
introduktion
effektiv befolkningsstørrelse (Ne) er en vigtig genetisk parameter, der estimerer mængden af genetisk drift i en population og er blevet beskrevet som størrelsen på en idealiseret Vrig–Fisher-population forventes at give den samme værdi af en given genetisk parameter som i den undersøgte population (krage og Kimura, 1970). Ne-størrelser kan påvirkes af udsving i folketællingsstørrelse (Nc), af avlskønforholdet og variansen i reproduktiv succes.
ne-estimering kan opnås ved hjælp af tilgange, der falder i tre metodologiske kategorier: demografisk, stamtavlebaseret eller markørbaseret (Flury et al., 2010). Stamtavle data er traditionelt blevet brugt til at opnå Ne estimater i husdyr. Imidlertid, pålidelige skøn over Ne afhænger af, at stamtavlen er komplet. Denne videnstilstand er mulig i nogle indenlandske befolkninger, hvis demografiske parametre er blevet nøjagtigt overvåget i et tilstrækkeligt stort antal generationer. I praksis forbliver anvendeligheden af denne tilgang imidlertid begrænset til nogle få tilfælde, der involverer stærkt administrerede racer (Flury et al., 2010; Uimari og Tapio, 2011).
en løsning til at overvinde begrænsningen af en ufuldstændig stamtavle er at estimere den nylige tendens i Ne ved hjælp af genomiske data. Flere forfattere har erkendt, at Ne kunne estimeres ud fra oplysninger om koblingsuligevægt (LD) (Sved, 1971; Hill, 1981). LD beskriver den ikke-tilfældige forening af alleler i forskellige loci som en funktion af rekombinationshastigheden mellem de fysiske positioner af loci i genomet. LD-signaturer kan dog også skyldes demografiske processer som blanding og genetisk drift (1943; 2005) eller gennem processer som “hitchhiking” under selektive fejninger (Smith og Haigh, 1974) eller baggrundsvalg., 1997). I sådanne scenarier bliver alleler på forskellige steder forbundet uafhængigt af deres nærhed i genomet. Forudsat at en population er lukket og panmiktisk, afhænger ld-værdien beregnet mellem neutrale ikke-linkede loci udelukkende af genetisk drift (Sved, 1971; Hill, 1981). Denne forekomst kan bruges til at forudsige Ne på grund af det kendte forhold mellem variansen i LD (beregnet ved hjælp af allelfrekvenser) og effektiv befolkningsstørrelse (Hill, 1981).
nylige fremskridt inden for genotypeteknologi (f. eks., ved hjælp af SNP-perlearrays med titusinder af DNA-prober) har muliggjort indsamling af store mængder genom-dækkende koblingsdata, der er ideelle til estimering af Ne hos blandt andet husdyr og mennesker (f.eks., 2007; De Roos et al., 2008; Corbin et al., 2010; Uimari og Tapio, 2011; Kijas et al., 2012). Imidlertid mangler et værktøj, der muliggør estimering af Ne fra LD, og forskere er i øjeblikket afhængige af en kombination af værktøjer til at manipulere data, udlede LD og har tendens til at bruge skræddersyede scripts til at udføre de relevante beregninger og estimere Ne.
Her beskriver vi SNeP, et programværktøj, der tillader estimering af Ne-tendenser på tværs af generation ved hjælp af SNP-data, der korrigerer for prøvestørrelse, fasning og rekombinationshastighed.
materialer og metoder
metoden SNeP bruger til at beregne LD afhænger af tilgængeligheden af fasede data. Når fasen er kendt, kan brugeren vælge Hill og Robertson (1968) kvadreret korrelationskoefficient, der gør brug af haplotype frekvenser til at definere LD mellem hvert par loci (ligning 1). I fravær af en kendt fase kan kvadreret Pearsons korrelationskoefficient for produktmoment mellem par af loci imidlertid vælges. Mens disse to tilgange ikke er de samme, er de meget sammenlignelige (McEvoy et al., 2011):
hvor pA og pB er henholdsvis frekvenserne af alleler A og B ved to separate loci (n−individer, pAB er frekvensen af haplotypen med alleler A og B i den undersøgte population, h og Y er de gennemsnitlige genotypefrekvenser for henholdsvis det første og det andet locus, HI er genotypen af individuel I ved det første locus, og Yi er genotypen af individuel I ved det andet locus. Ligning (2) korrelerer de genotypiske alleltællinger i stedet for haplotypefrekvenserne og påvirkes ikke af dobbelt heterosygoter (denne tilgang resulterer i de samme estimater som –r2-indstillingen i PLINK).
SNeP estimerer den historiske effektive befolkningsstørrelse baseret på forholdet mellem r2, Ne og c (rekombinationshastighed), (ligning 3—Sved, 1971) og gør det muligt for brugerne at medtage korrektioner for stikprøvestørrelse og usikkerhed i den gametiske fase (ligning 4—overløb og bakke, 1980):
hvor n er antallet af individuelle samplede, larr = 2, når den gametiske fase er kendt, og larr = 1, hvis fasen i stedet ikke er kendt.
flere tilnærmelser bruges til at udlede rekombinationshastigheden ved hjælp af den fysiske afstand (kr) mellem to loci som reference og oversætte den til koblingsafstand (d), som normalt beskrives som Mb(kr) KRP cM(d). For små værdier af d er sidstnævnte tilnærmelse gyldig, men for større værdier af d øges sandsynligheden for flere rekombinationshændelser og interferens, desuden er forholdet mellem kortafstand og rekombinationshastighed ikke lineær, da den maksimale rekombinationshastighed er 0,5. Således, medmindre du bruger meget kort KRP, er tilnærmelsen d KRP ikke ideel (Corbin et al., 2012). Vi implementerede derfor kortlægningsfunktioner for at oversætte det estimerede d til c efter Haldane (1919), Kosambi (1943), Sved (1971) og Sved og Feldman (1973). I første omgang udleder SNeP d for hvert par SNP ‘ er som direkte proportional med KRP ifølge d = K-KRP, hvor k er en brugerdefineret rekombinationshastighedsværdi (standardværdi er 10-8 som i Mb = cM). Den udledte værdi af kurp kan derefter underkastes en af de tilgængelige kortlægningsfunktioner, hvis det kræves af brugeren.
løsning af ligning (3) For Ne og inklusive alle de beskrevne korrektioner tillader forudsigelse af Ne fra LD-data ved hjælp af (Corbin et al., 2012):
hvor Nt er den effektive populationsstørrelse t generationer siden beregnet som t = (2F(ct))-1 (Hayes et al., 2003), ct er rekombinationshastigheden defineret for en specifik fysisk afstand mellem markører og eventuelt justeret med kortlægningsfunktionerne nævnt ovenfor, r2adj er LD-værdien justeret for prøvestørrelse og Kur:= {1, 2, 2.2} er en korrektion for forekomsten af mutationer (Ohta og Kimura, 1971). Derfor, LD over større rekombinante afstande er informativ om nylige Ne, mens kortere afstande giver information om fjernere tider i fortiden. Et binning-system implementeres for at opnå gennemsnitlige r2-værdier, der afspejler LD for specifikke inter-locus-afstande. Det implementerede binning-system bruger følgende formel til at definere minimums – og maksimumsværdierne for hver bin:
hvor bi (kr1) er Ith bin af det samlede antal skraldespande (totBins), sind og maks er henholdsvis minimum og maksimum afstanden mellem SNP ‘ er og K er et positivt reelt tal (kr0), når k er lig med 1, fordelingen af afstande mellem skraldespandene er lineær, og hver bin har samme afstandsområde. For Større værdier ændres fordelingen af afstande, hvilket giver et større interval på de sidste skraldespande og et mindre interval på de første skraldespande. Ved at variere denne parameter kan brugeren have et tilstrækkeligt antal parvise sammenligninger til at bidrage til det endelige ne-estimat for hver bin.
eksempel ansøgning
Vi testede SNeP med to offentliggjorte datasæt, der tidligere var blevet brugt til at beskrive tendenser i Ne over tid ved hjælp af LD, Bos indicus og Ovis aries . R2-estimaterne for kvægdatasætene blev opnået af forfatterne ved hjælp af GenABLE (aulchenko et al., 2007) ved hjælp af en minimum allelfrekvens (MAF) < 0,01 og justering af rekombinationshastigheden ved hjælp af Haldanes kortlægningsfunktion (Haldane, 1919). R2-estimaterne af fåredataene blev beregnet af forfatterne ved hjælp af PLINK-1.07 (Purcell et al., 2007), med en MAF < 0.05 og ingen yderligere rettelser. For begge autosomale datasæt R2 estimater, hvor korrigeret for stikprøvestørrelse ved hjælp af ligning (4) med LR = 2. Til disse sammenlignende analyser inkluderede SNeP-kommandolinjen de samme parametre, der blev brugt til de offentliggjorte data bortset fra R2-estimaterne, beregnet gennem genotypetælling og brugen af Sneps nye binning-strategi.
resultater
SNeP er en flertrådet applikation udviklet i C++ og binære filer til de mest almindelige operativsystemer (f.eks. De binære filer ledsages af en manual, der beskriver den trinvise brug af SNeP til at udlede tendenser i Ne som beskrevet her. SNeP producerer en outputfil med tabulatorafgrænsede kolonner, der viser følgende for hver bin, der blev brugt til at estimere Ne: antallet af generationer i fortiden, som bin svarer til (f. eks., 50 generationer siden), det tilsvarende ne-estimat, den gennemsnitlige afstand mellem hvert par SNP ‘er i skraldespanden, den gennemsnitlige r2 og standardafvigelsen for r2 i skraldespanden og antallet af SNP’ er, der bruges til at beregne r2 i skraldespanden. Denne fil kan nemt importeres i Microsoft R eller andre programmer til at plotte resultaterne. De her viste tomter (Figur 1, 3) svarer til kolonnerne for generationer siden og Ne fra outputfilen. Kolonnen med R2-standardafvigelsen er til rådighed for brugerne at inspicere variansen i Ne-estimatet i hver skraldespand, især for de skraldespande, der afspejler ældre tidsestimater, og som er mindre pålidelige, da antallet af SNP ‘ er, der bruges til at estimere r2, bliver mindre.
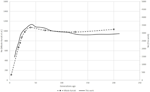
Figur 1. Sammenligning af Ne-tendenser for seks Svenske fårracer ifølge Burren et al. (2014) (stiplede linjer) og dette arbejde (solide linjer).
det format, der kræves til inputfilerne, er standard PLINK-format (ped og kortfiler) (Purcell et al., 2007). SNeP giver brugerne mulighed for enten at beregne LD på dataene som beskrevet ovenfor eller bruge en brugerdefineret forudberegnet LD-matrice til at estimere Ne ved hjælp af ligning (5).programgrænsefladen giver brugeren mulighed for at kontrollere alle parametre i analysen, f.eks. afstandsområdet mellem SNP ‘ er i bp og det sæt kromosomer, der anvendes i analysen (f. eks. 20-23). Derudover inkluderer SNeP muligheden for at vælge en MAF-tærskel (standard 0.05), da det har vist sig, at regnskabsmæssig behandling af MAF resulterer i upartiske R2-estimater uanset stikprøvestørrelse (Sved et al., 2008). Sneps flertrådede arkitektur tillader hurtig beregning af store datasæt (vi testede op til ~100k SNP’er for et enkelt kromosom), for eksempel blev de her beskrevne BOS-data analyseret med en processor i 2’43”, brugen af to processorer reducerede tiden til 1’43”, fire processorer reducerede analysetiden til 1 ’05”.
Sebu eksempel
for Sebu-analysen viste formerne af Ne-kurverne opnået med SNeP og deres offentliggjorte datatendenser den samme bane med et glat fald indtil for omkring 150 generationer siden, efterfulgt af en udvidelse med en top for omkring 40 generationer siden og sluttede i et stejlt fald på de seneste generationer (Figur 1). Mens tendenserne i begge kurver imidlertid var de samme, resulterede de to tilgange i forskellige Ne-estimater, hvor Sneps værdier var cirka tre gange større end dem i det originale papir. Mens vi forsøgte at bruge forfatterens parametre i vores analyser, var nogle forskelle uundgåelige, dvs.den oprindelige offentliggørelse af kvægdata estimeret r2 med en anden tilgang til den, der blev implementeret i SNeP. Analyser med SNeP var baseret på genotyper, mens den oprindelige analyse var baseret på udledte to locus haplotyper, hvilket resulterer i de offentliggjorte data, der viser en forventet r2 på 0,32 ved minimumsafstanden, mens vores estimater var 0,23. Tilsvarende Mbole-Kariuki et al. (2014) opnåede et baggrundsniveau r2 = 0,013 omkring 2 Mb, mens vores estimat i samme afstand var 0.0035 (data ikke vist). Følgelig, da vores estimater af LD var konsekvent mindre end Mbole-Kariuki et al. (2014) Det forventes, at vores Ne-estimater skal være større. Mens denne observation fremhæver vigtigheden af et omhyggeligt valg af parametrene og deres tærskler, er det vigtigt at fremhæve, at selv om den absolutte størrelse af Ne-værdierne er forskellig, er tendenserne næsten identiske.
Sveriges Fåreeksempel
de seks Sveriges fåreracer analyseret med SNeP producerede sammenlignelige resultater med dem fra det originale papir (figur 2) med for det meste overlappende ne-trendkurver (figur 3). Den generelle tendens i Ne viste imidlertid et fald mod nutiden. SNeP producerede lidt større værdier af Ne for den fjernere fortid (700-800 generationer). Dette skyldes det forskellige binning-system, der anvendes i SNeP, som giver brugeren mulighed for at opnå en mere jævn fordeling af parvise sammenligninger inden for hver bin (dvs., antallet af SNP parvise sammenligninger inden for hver bin er sammenlignelig). For tidsrummet, der strækker sig ud over 400 generationer siden, Burren et al. (2014) brugte kun tre skraldespande i deres analyse (centreret ved 400, 667 og 2000 generationer siden), mens SNeP i samme tidsrum brugte 5 skraldespande med et antal parvise sammenligninger afhængigt af det interval,der er defineret med formler 6a, b. derfor ender Burren og kollegers tilgang med en højere tæthed af data, der beskriver de seneste generationer end at beskrive de ældste generationer. Derfor har brugen af færre skraldespande en tendens til at øge tilstedeværelsen af mindre værdier af Ne i hver skraldespand og følgelig sænke den gennemsnitlige Ne-værdi for hver skraldespand. Ne-værdierne for den nylige fortid sammenlignet med den 29. generation i fortiden gav meget lignende resultater. Den største forskel (50) blev opnået for SBS-racen.

figur 2. Sammenligning mellem nylige Ne-værdier beregnet ved 29. generation i dette arbejde og Burren et al. (2014) for seks Svenske fåreacer.
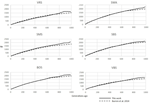
figur 3. Sammenligning af Ne-tendenser for de sidste 250 generationer i de data, der er opnået af Mbole-Kariuki et al. (2014) (stiplet linje) og ved hjælp af SNeP (solid linje).
Diskussion
analyse af Ne ved hjælp af LD-data blev først demonstreret for 40 år siden og er blevet anvendt, udviklet og forbedret siden (Sved, 1971; Hayes et al., 2003; Tenesa et al., 2007; De Roos et al., 2008; Corbin et al., 2012; Sved et al., 2013). Det traditionelt lille antal analyserede SNP ‘er er ikke længere en begrænsning, da SNP-Chips omfatter et ekstremt stort antal SNP’ er, der er tilgængelige på kort tid og til en rimelig pris. Dette har øget brugen af metoden, som er blevet anvendt på mennesker (Tenesa et al., 2007; McEvoy et al., 2011) såvel som til flere tamme arter (England et al., 2006; Uimari og Tapio, 2011; Corbin et al., 2012; Kijas et al., 2012). Sammen med disse forbedringer, metodologiske begrænsninger er blevet tydelige og er blevet behandlet her, med størstedelen af indsatsen, der peger på den korrekte estimering af den nylige Ne. Alligevel er den kvantitative værdi af estimatet meget afhængig af stikprøvestørrelse, typen af LD-estimering og binningsprocessen (Vaples and Do, 2008; Corbin et al., 2012), mens dets kvalitative mønster afhænger mere af den genetiske information end af datamanipulation.
indtil videre er denne metode blevet anvendt ved hjælp af en række programmer, der findes ingen standardiseret tilgang til bin resultaterne, og hver undersøgelse har anvendt en mere eller mindre vilkårlig tilgang, f.eks., 2012), binning for fjernklasser med et konstant interval for hver bin (Kijas et al., 2012) eller binning pr.fjernklasser lineært, men med større skraldespande til de nyere tidspunkter (Burren et al., 2014). Så vidt vi ved, er det eneste tilgængelige program, der estimerer Ne gennem LD, NeEstimator (Do et al., 2014), en opgraderet version af den tidligere LDNE (Vaples and Do, 2008), der tillader analyse af stort datasæt (som 50k SNPChip). Det er vigtigt, at mens SNeP fokuserer på at estimere historiske Ne-tendenser, er Neestimators mål at producere nutidige upartiske Ne-estimater, sidstnævnte bør derfor betragtes som et komplementært værktøj, mens man undersøger demografi gennem LD.
Vi brugte SNeP til at analysere to datasæt, hvor metoden tidligere blev anvendt. De resultater, vi opnåede for fårdataene, var både kvantitativt og kvalitativt sammenlignelige med dem, der blev opnået af Burren et al. (2014), mens vi for EBU-dataene opnåede et ne-trendestimat, der nøje matchede Mbole-Kariuki et al. (2014) selvom vores punktestimater af Ne var større end dem, der er beskrevet for dataene (Mbole-Kariuki et al., 2014). Uoverensstemmelsen mellem disse to resultater afspejler, at Burren og kolleger producerede deres R2-estimater ved hjælp af PLINK (standardprogrammet til storskala SNP-datamanipulation), der bruger den samme tilgang, der blev brugt til at estimere R2 af SNeP, mens Mbole-Kariuki et al. fulgte Hao et al. (2007) til R2 estimering. Brugen af forskellige estimater for LD er kritisk for det kvantitative aspekt af Ne-kurven, hvor på grund af den hyperbolske korrelation mellem Ne og r2, et fald i r2 på dets interval tættere på 0 kan føre til en meget stor ændring i Ne-estimater, mens forskelle i estimater er mindre signifikante, når r2-værdien er høj, dvs.tættere på 1. Selvom Ne-værdierne i et af datasætene var væsentligt forskellige, overlappede Ne-kurverne i begge tilfælde med dem, der oprindeligt blev offentliggjort.
som allerede foreslået af andre forfattere skal pålideligheden af de kvantitative estimater opnået med denne metode tages med forsigtighed, især for Ne-værdier relateret til de nyeste og ældste generationer (Corbin et al., 2012) fordi for de seneste generationer er store værdier af c involveret, hvilket ikke passer til de teoretiske implikationer, som Hayes foreslog at estimere en variabel Ne over tid (Hayes et al., 2003). Estimater for de ældste generationer kan også være upålidelige, da coalescent teori viser, at ingen SNP kan samples pålideligt efter 4Ne generationer i fortiden (Corbin et al., 2012). Yderligere, ne-estimater, og især dem, der er relateret til generationer længere i fortiden, påvirkes stærkt af datamanipulationsfaktorer, såsom valget af MAF-og alfaværdier. Derudover kan den anvendte binningsstrategi forstyrre metodens generelle præcision, for eksempel hvor et utilstrækkeligt antal parvise sammenligninger bruges til at udfylde hver bin.
en af anvendelserne af metoden er at sammenligne racedemografier. I dette tilfælde ville formen på Ne-kurverne være det optimale værktøj til at differentiere forskellige demografiske historier mere end deres numeriske værdier ved at bruge dem som et potentielt demografisk fingeraftryk for den race eller art, men under hensyntagen til, at mutation, migration og udvælgelse kan påvirke Ne-estimeringen gennem LD (Vaples and Do, 2010). Derudover er omhyggelig overvejelse af de data, der analyseres med SNeP (og andre programmer til estimering af Ne), meget vigtig, da tilstedeværelsen af forvirrende faktorer som blanding kan resultere i partiske estimater af Ne (2014).
formålet med SNeP er derfor at give et hurtigt og pålideligt værktøj til at anvende LD-metoder til at estimere Ne ved hjælp af genotypiske data med høj kapacitet på en mere konsistent måde. Det giver mulighed for to forskellige R2 estimeringsmetoder plus muligheden for at bruge r2 estimater fra eksterne programmer. Brugen af SNeP overvinder ikke grænserne for metoden og teorien bag den, men det giver brugeren mulighed for at anvende teorien ved hjælp af alle korrektioner, der er foreslået til dato.
Forfatterbidrag
MB udtænkt og skrev programmet og manuskriptet. MB, MT og Hf testede programmet og udførte analyserne. MF, MF og MF har revideret manuskriptet. Alle forfattere godkendte det endelige manuskript.
interessekonflikt Erklæring
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
anerkendelser
Vi takker Christine Flury for at have leveret fårdataene og for nyttig diskussion. Vi takker også de to korrekturlæsere for nyttige forslag til forbedring af dette papir. MB blev støttet af programmet Master og Back (Regione Sardegna).(1997). Virkningerne af lokal selektion, afbalanceret polymorfisme og baggrundsvalg på ligevægtsmønstre for genetisk mangfoldighed i underinddelte populationer. Genet. Res. 70, 155-174. doi: 10.1017 / S0016672397002954
PubMed abstrakt | Fuld tekst | CrossRef Fuld tekst/Google Scholar
krage, J. F. og Kimura, M. (1970). En introduktion til Populationsgenetikteori. Harper og række.
Google Scholar
Ohta, T. og Kimura, M. (1971). Koblingsuligevægt mellem to segregerende nukleotidsteder under den stadige strøm af mutationer i en endelig population. Genetik 68, 571-580.
PubMed abstrakt / Fuld tekst | Google Scholar
S. (1943). Isolering efter afstand. Genetik 28, 114-138.
PubMed abstrakt / Fuld tekst | Google Scholar